Help for using XDock server
1. How to provide input for docked molecules
The XDock server is for predicting the complex structure between a protein/nucleic-acids and a small molecular though a distance geometric sampling method and a iterative score function (ITScore/ITScoreNL). Users need to provide inputs for the protein and molcules to be docked.
- Upload your pdb file in PDB format or Mol2 format or CIF format.
1.1 provide receptor for docking
The XDock server is supports multiple input methods. The first is the most common file input. The user can click the "select file" button and then select the file to be uploaded.
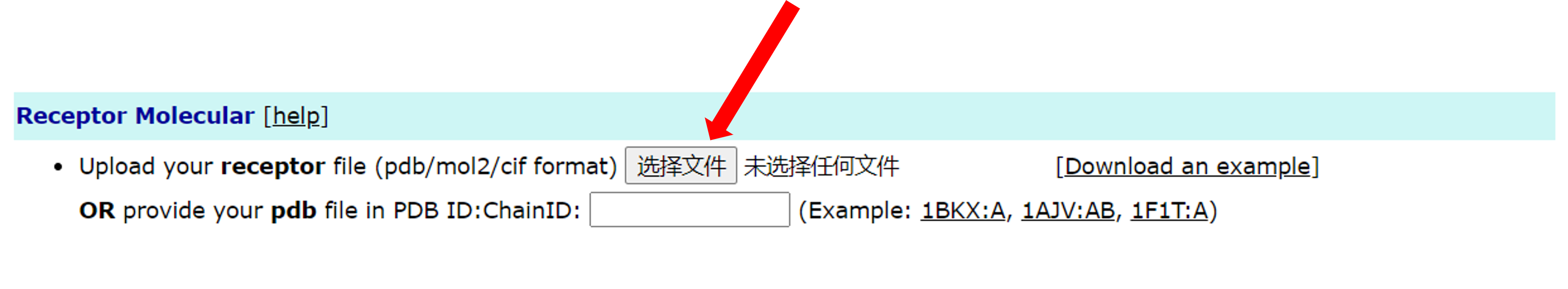
The second is to enter the serial number of PDB and the chain. Users can enter the ID of the PDB and the corresponding chain ID in the text box. For example, like 1BKX:A:
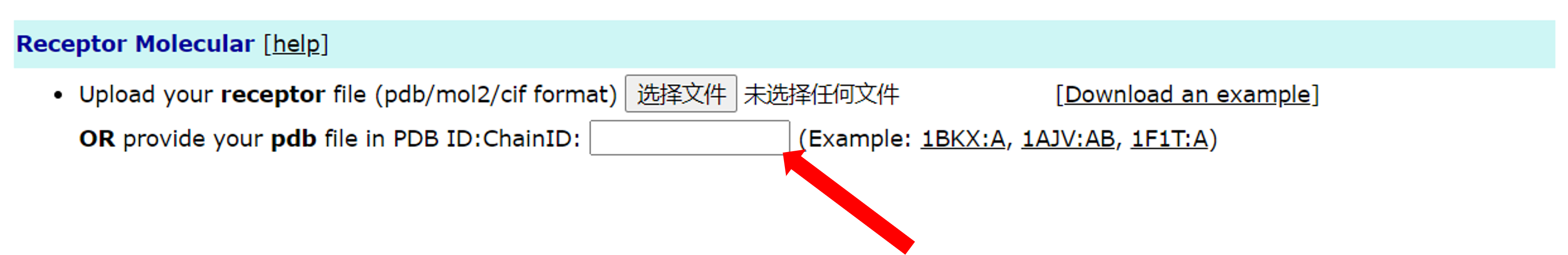
If the user enters the file and PDB ID at the same time, the priority of the file will be higher.
1.2 provide ligand for docking
For ligands, the XDock server also supports multiple ways. The first is the normal file input, which supports files in formats such as PDB, MOL2, SDF formats.
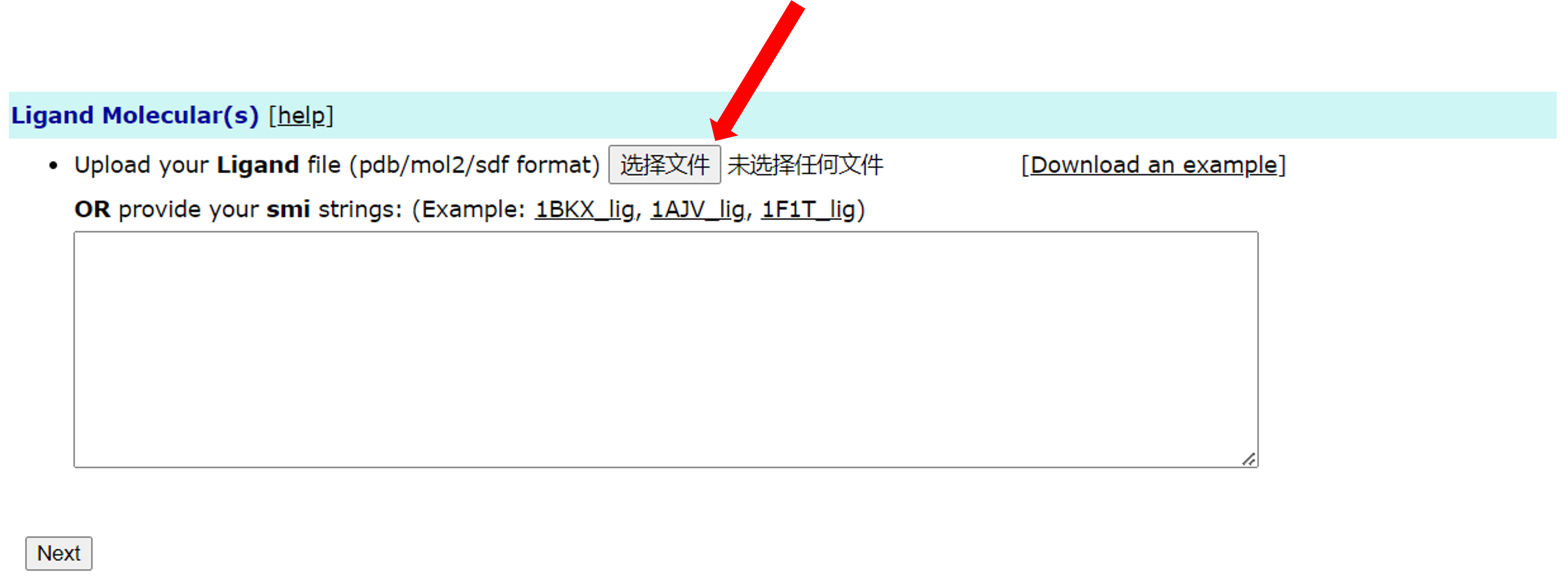
The second is the input of the SMILES string. Users can enter the SMILES string of the ligand in the text box below. Such as the following (Users can also click 1BKX_lig in the Example to directly fill in the text box):
OC[C@H]1O[C@H]([C@@H]([C@@H]1O)O)n1cnc2c(N)ncnc12 1BKX_ligand
Among them, the first column is SMILES string, and the second column is the name of the ligand.
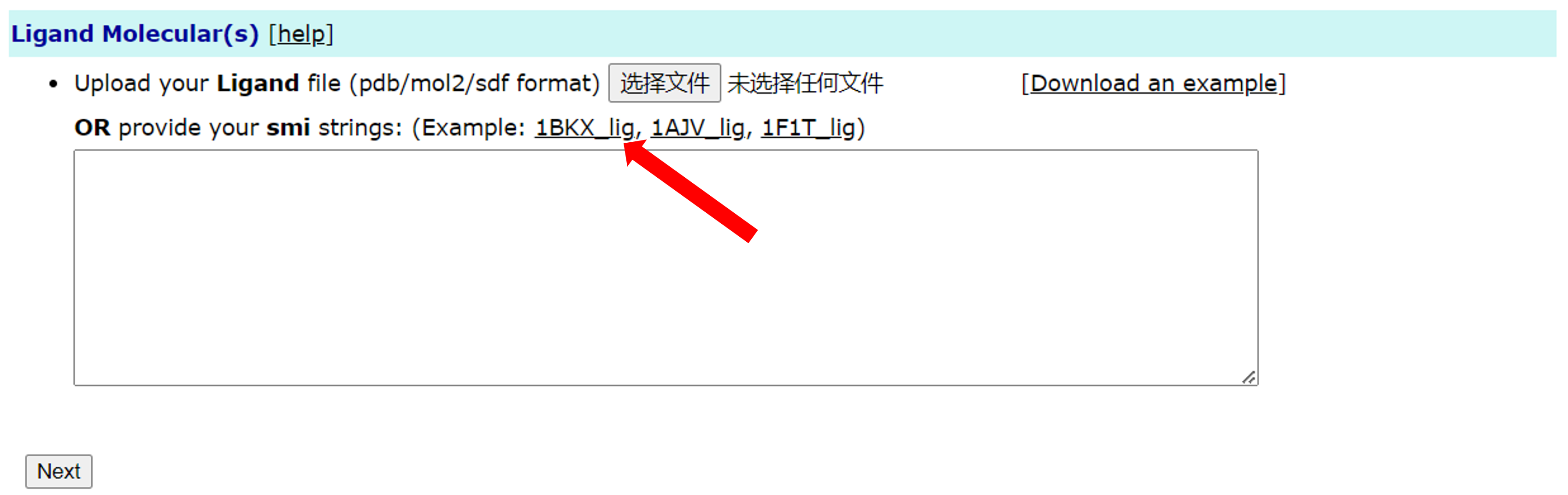
If the user enters the file and PDB ID at the same time, the priority of
the file will be higher.
Then the user can click Next to enter the next interface.
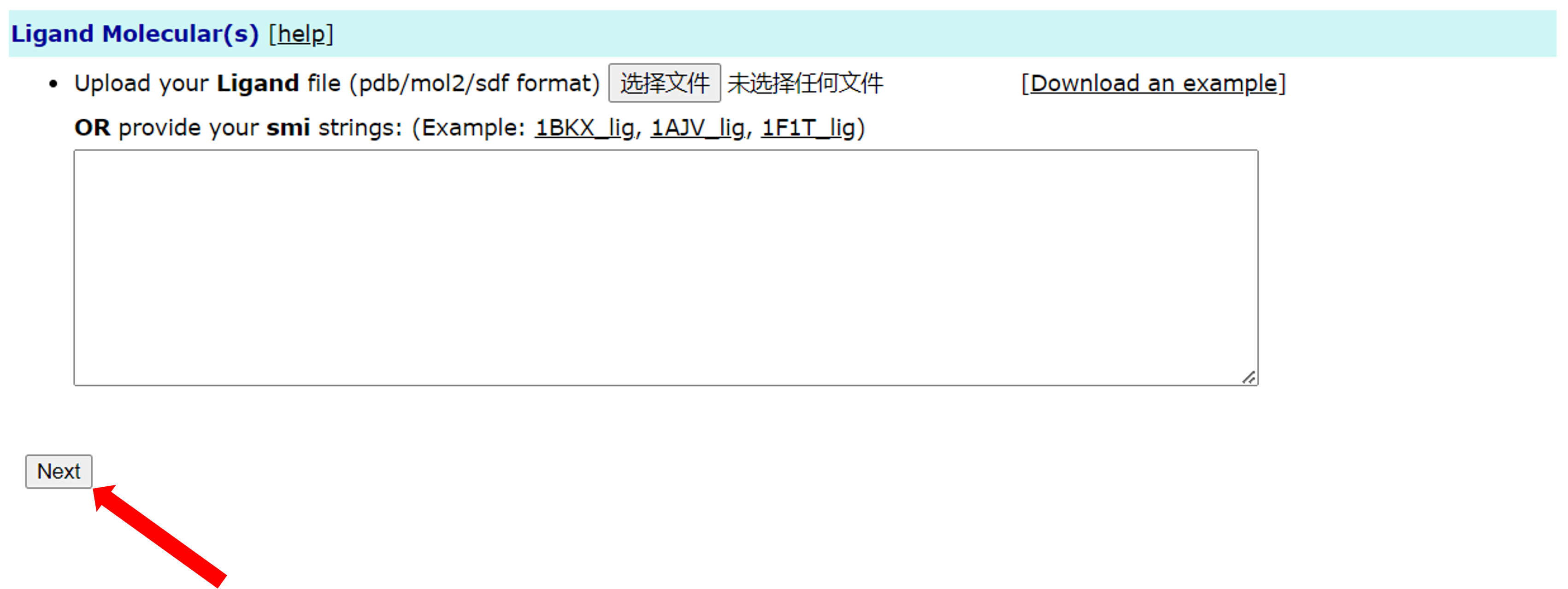
1.3 select site for docking
After clicking "Next", the user enters the interface of the selection of the site. First of all, it is necessary to wait for a few seconds to one minute to calculate the binding site. When the interface of the loading is over, the user can see the three-dimensional structure of the receptor and the first binding pocket selected by the default.
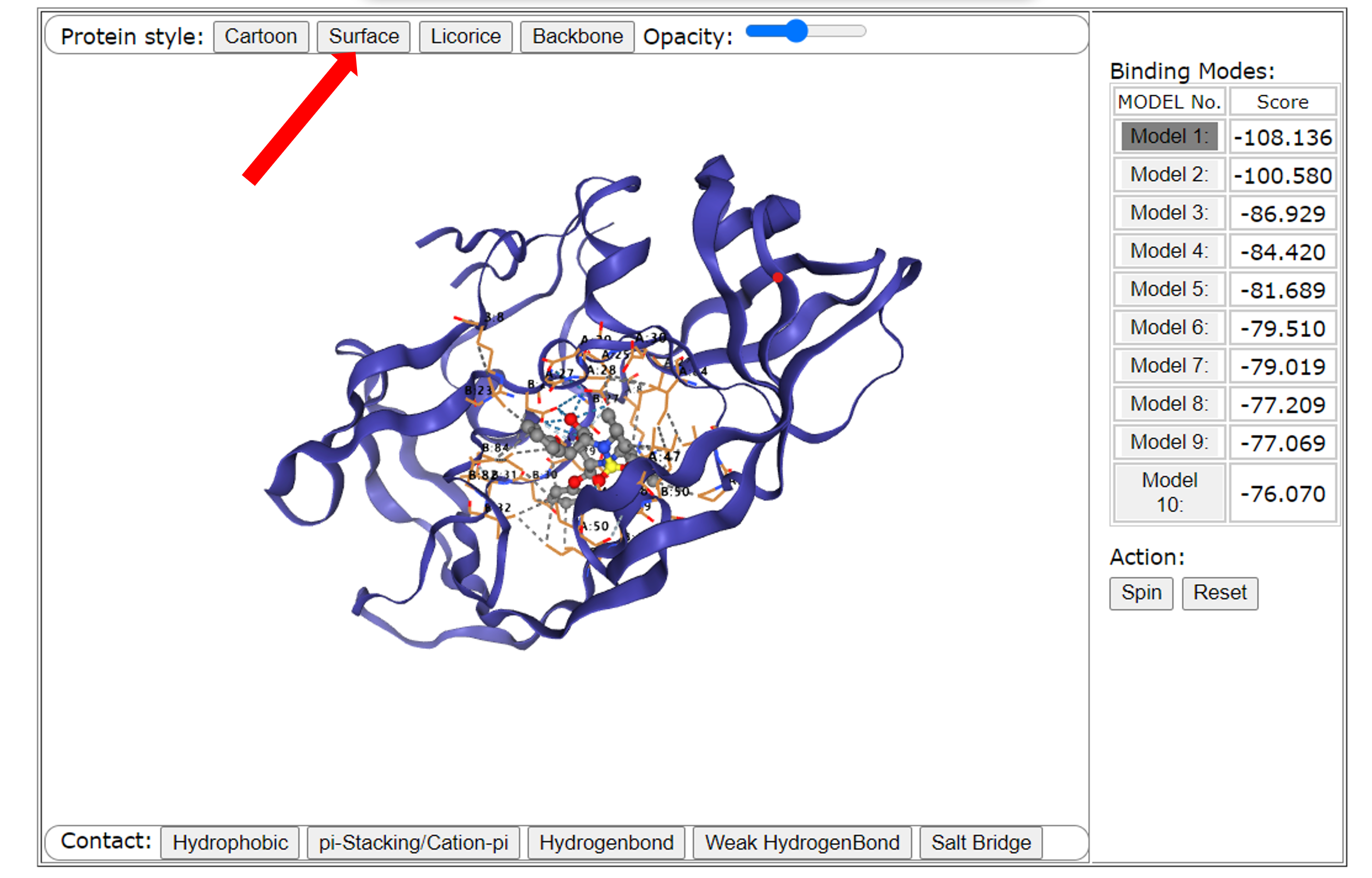
Users can use the mouse to change the perspective and view the receptor and pocket. Users can change the size of the viewing angle (enlarge or reduce the viewing angle) with the mouse wheel. Users can also control the protein display mode through the three buttons on the upper right: Cartoon, Surface, and Licorice, and restore the original viewing angle through reset, and adjust the transparency of the protein surface through Opacity.
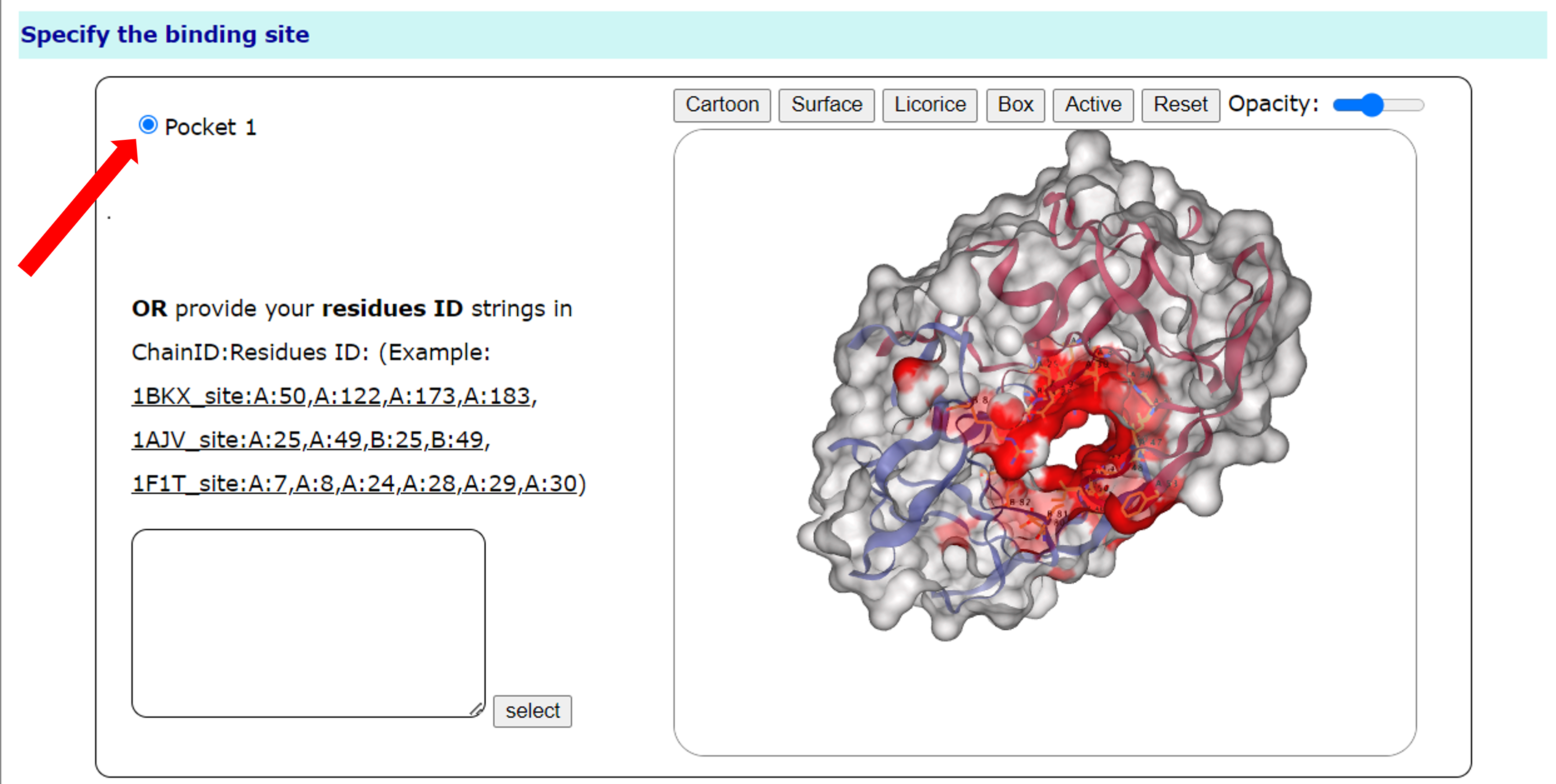
Users can choose different pocketsin the options on the left side of the interface. Of course, users can also select the residual base of the binding site in the text box below, for example
A:50,A:122,A:173,A:183
Then click the select button to select the amino acid serial numbe entered by the user as a binding site.
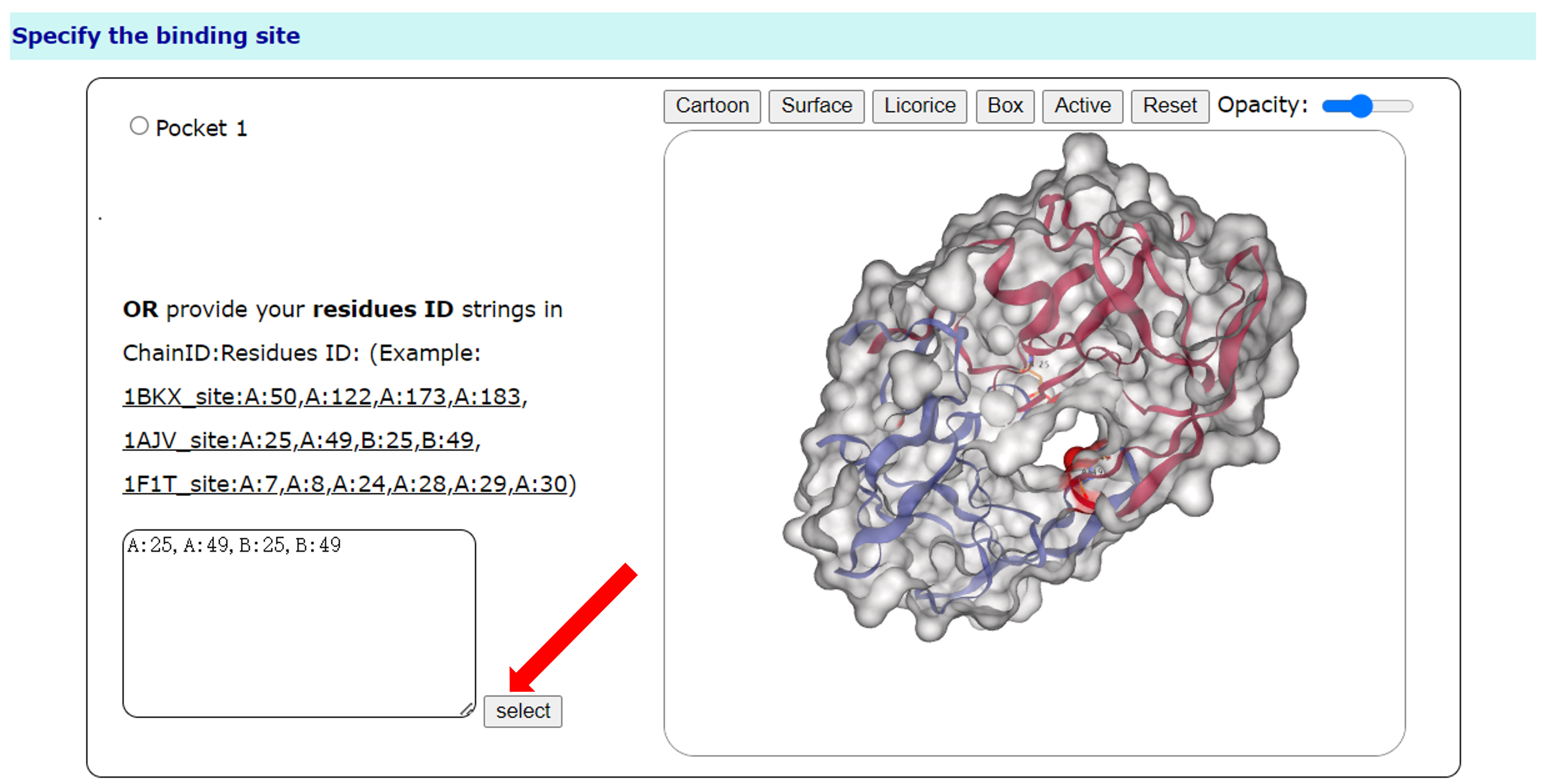
It should be emphasized that users can only choose one in the "pocket" and input sites. If the user clicks any "pocket", the remaining pocket and input site will be abandoned automatically. If the user chooses an input site, then all pocket will be abandoned.
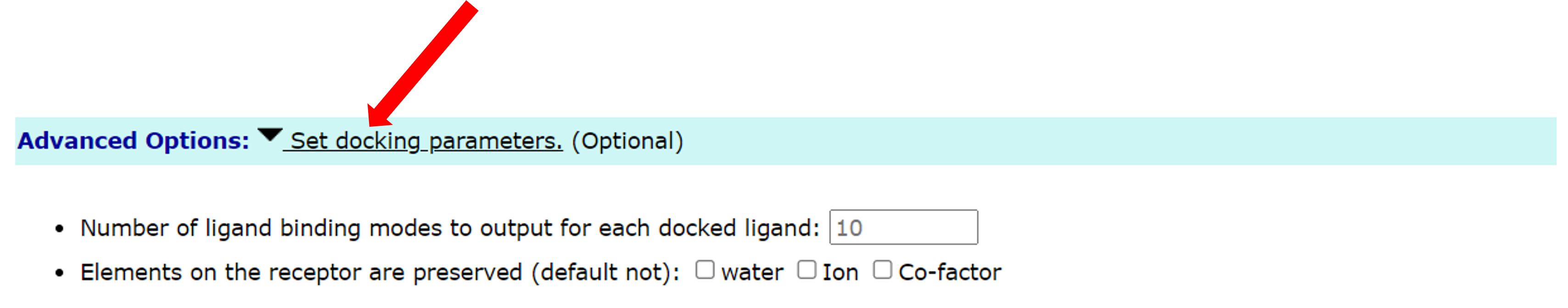
1.4 advanced options for docking
Users can also make additional settings in Advanced Options. Including two options:
- 1. The number of ligand binding modes to output for each docked ligand, the default value is 10.
- 2. Select whether to keep water molecules or ions or co-factor, the default is not retained.
Finally, the user can click "submit" to submit the task and wait a little after a while to get the result. Users can fill in the mailbox address. If the dock is completed, XDock server will send an email to notify you.

2. How to obtain your XDock results.
Once users submit their job, they will be redirected to a status web page showing the status of the job. The status page is automatically refreshed every 10 seconds until the job is finished. Users have three ways to obtain their docking results.
- Keep the status page open until it shows the docking results when the job is finished.
- Bookmark the status page and come back later to check the docking results.
- Wait for the email notification if users provide a valid email address when they submit their job.
After the job is done, users will be redirected to the result page, from which they can download the following files
- Receptor Mol2 file converted from a user uploaded receptor file.
- The individual ligand models of the top 10 predictions that were docked to the receptor.
- The compressed packages for the top 10 binding models or all the docking results.
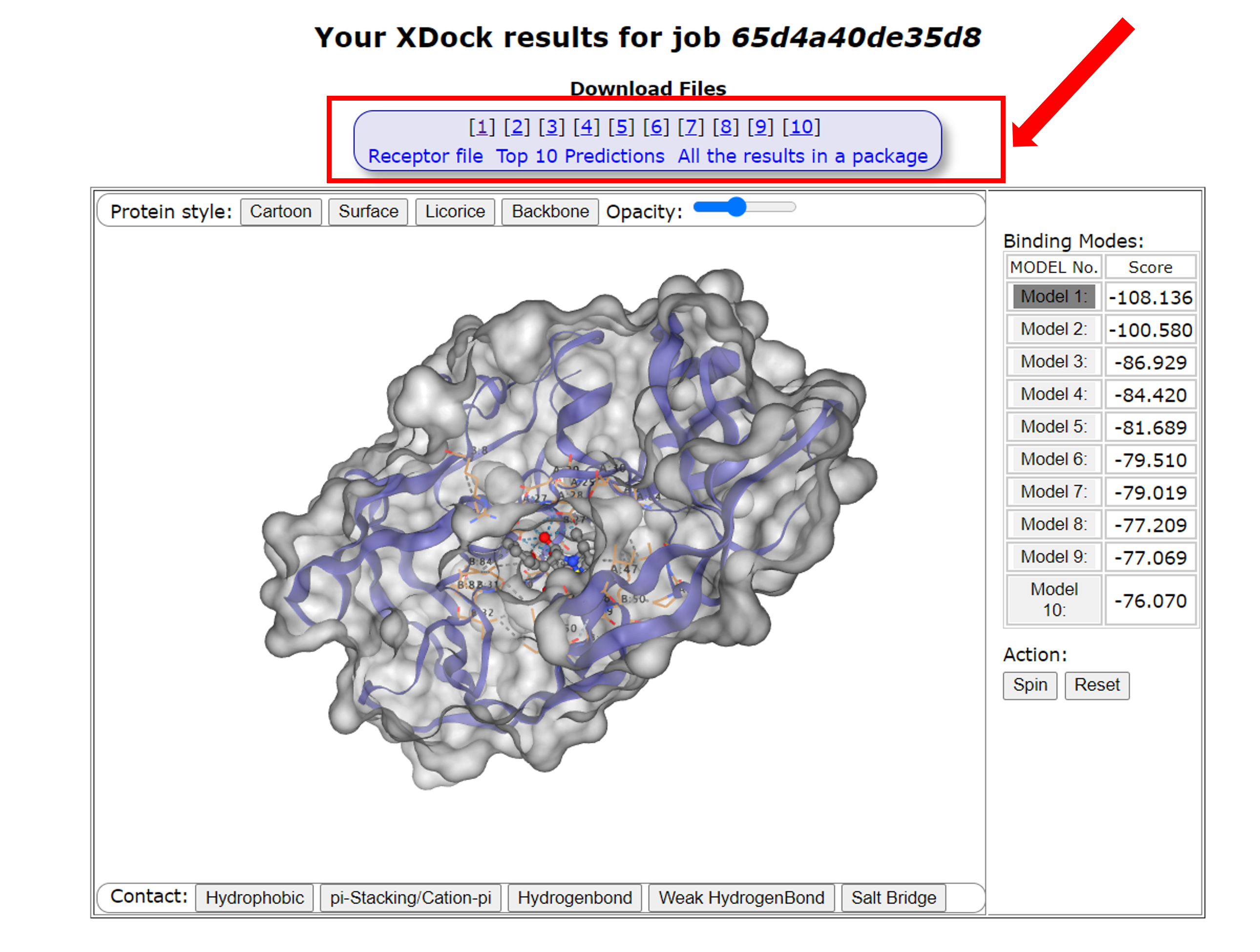
Since the top 10 binding models are normally deemed as the most important models, the result page also provides an interactive view of the top 10 models using the ngl software. Users can choose to view any of the top 10 models or all together.
The page also gives a summary of the rankings and docking scores for the top 10 binding models.
3. Detailed description of the docking results of XDock server.
The content of the file download has been described above. The following is an introduction to the interface display interface. Users can click the "Output Example" button on the XDock homepage to view the output example.
First, we will display the binding mode of 10 ligands and receptors on the interface. Users can select different binding modes to view the results in the "Model" button on the right. If the user clicks a certain "Model", the button will change from white to gray, and the binding mode will also be displayed in the interface. The value corresponding to the binding mode is also displayed on the right side of the button. Users can also use the "spin" button for automatic rotation of the perspective, and "reset" to reset perspective.
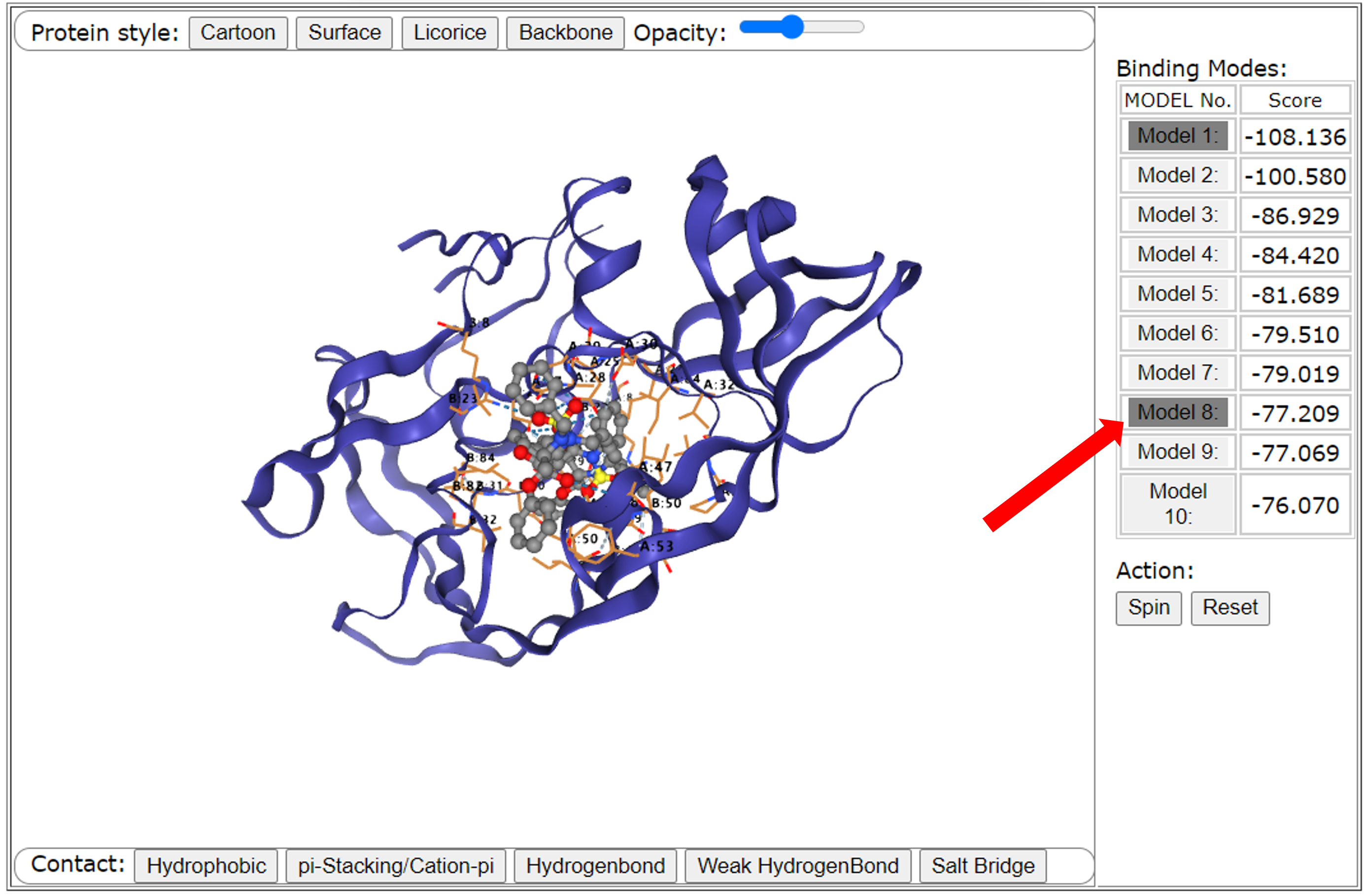
Second, we will briefly introduce how to use this graphical interface. Users can use the mouse to change the perspective. The left button of the mouse can rotate the molecule. Hold down the right mouse button to drag the perspective. Slide the mouse wheel to change the size of the vision.
Third, users can change the display style of the protein through a row of buttons above the interface. These buttons correspond to the four display status of the receptor from left to right: "Cartoon", "Surface", "Licorice", and "Backbone". Among them, "Cartoon" and "Surface" are selected by default. Users can also dynamically control the transparency of "Surface" through a "Opacity Slider".
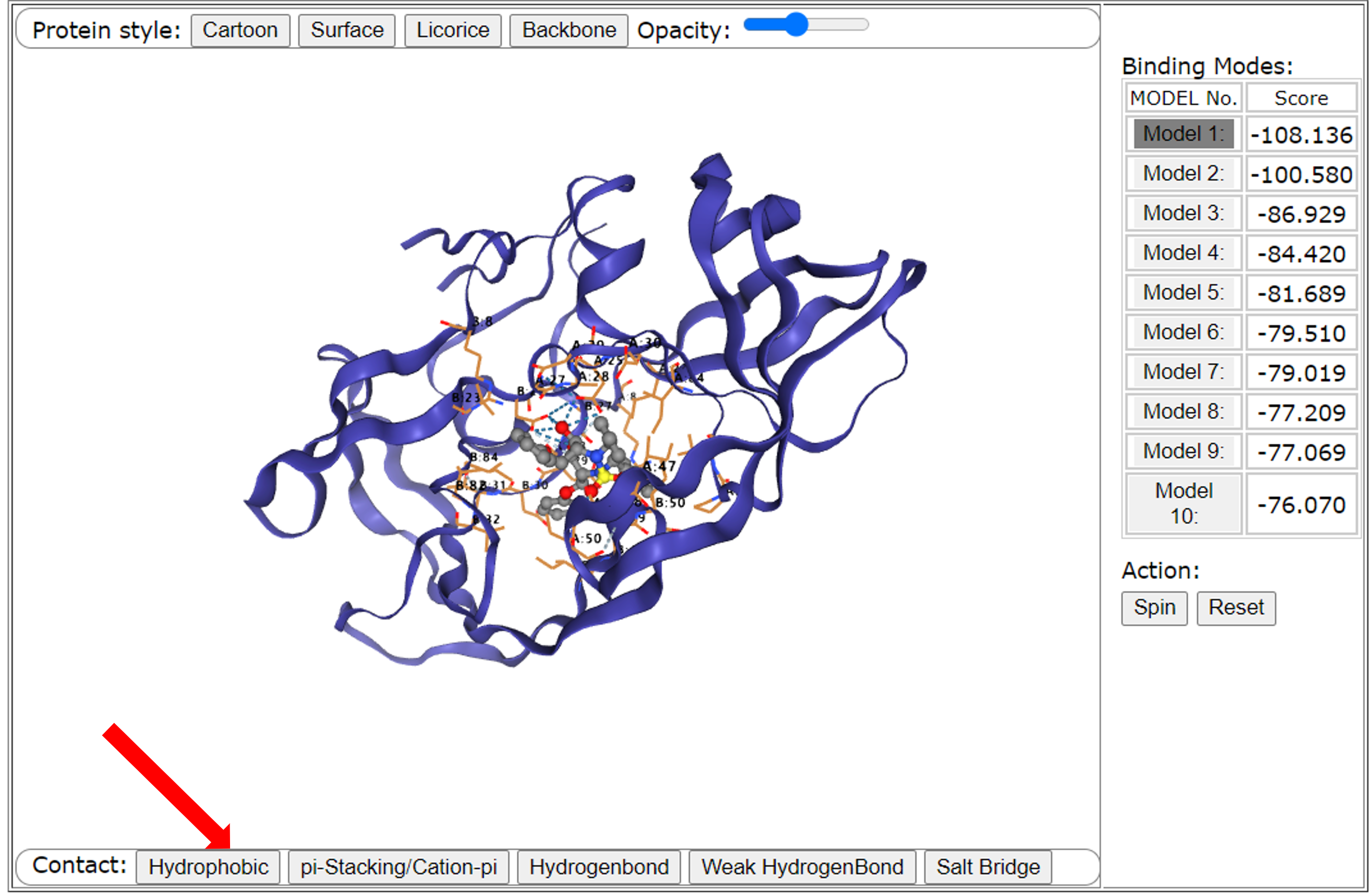
Finally, users can control whether to display some interactions between the ligand and the receptor through the row of the button below the interface. We take hydrophobic interaction as an example. If there is a hydrophobic interaction between the receptor and the ligand, we can control whether it shows it through the "Hydrogen" button, and it is displayed by default. Similarly, we can control the display of interactions between aromatic rings (ions) on the interface by using the "pi-Stacking/Cation-pi" button, we can also show hydrogen bond interactions by using the "Hydrogen Bond" button, weak hydrogen bond interactions (halogen bonding) can be displayed through the "Weak Hydrogenbond'" button; and salt bridge interactions can be shown by using the "Salt Bridge" button.
4. How to view the docking results on the local.
The first step is to decompresses the downloaded all_results.tar.gz package.
In the second step, users uses the visualization software
UCSF Chimera
to view the results of the docking as follows:
- Click the File button in the toolbar and select the open option from the drop-down menu.
- Select the xdock_receptor.mol2 file in the folder you extracted.
- Click the Tools button in the toolbar and select ViewDock from the Surface/Binding Analysis option in the drop-down menu.
- Select the xdock_ligands.mol2 file in the folder you extracted.
- Select the Dock 4, 5 or 6 option in the window that pops up.
- The window that pops up contains information about all the docking conformations, and users can select different items to view conformation.
- Users can press and hold the window with the left side of the mouse to change the viewing Angle, or zoom in or out with the wheel
- Users can refer to UCSF Chimera's Documentation for more information on how to operate.