Introduction of EM2NA
EM2NA is developed to build nucleic-acids (NA) structure from cryo-EM maps. The workflow of EM2NA is illustrated in Figure 1. EM2NA starts with the detection of DNA/RNA regions in a raw cryo-EM map through a Swin-Conv-UNet (SCUNet) architecture that has simultaneous local and nonlocal learning capabilities. The stage-1 network is trained to segment a cryo-EM map into NA, protein, and background. After the segmentation, the DNA/RNA density region is use as input of the stage-2 network to predict the backbone atom probabilities and nucleotide types. The two networks adopt the same SCUNet architecture and only differ in the output channels. The neural networks are trained with 322 experimental density maps. The backbone atom probabilities output from the stage-2 network are then converted to three-dimensional (3D) points by detecting the local maxima using a mean-shift algorithm. Next, the combined P and C4' points are traced to multiple backbone paths by solving a Vehicle Routing Problem (VRP). For each path, the C4' points are first extracted and the P points are placed for every two C4' atoms to form a "...(C4'-P-C4')..." order. The N1 or N9 points are then assigned for each C4' point by querying the first-nearest neighbor. In addition, two more P points are added at both terminals. The direction of each path can be easily determined by considering the nucleotide geometries. After the path direction determination, we utilize the Smith-Waterman algorithm to assign the sequence for each of the built backbones. The sequence assignment is further improved by considering the base pairing between nucleotides in double helices. With the backbone path and assigned nucleotide types, the full-atom structure is built by aligning template nucleotide conformations onto the P-C4'-N1/N9 backbone using the Kabsch algorithm. Optionally, a post-refinement can be performed to relax the structure and improve side-chain conformations and map-model correlations.
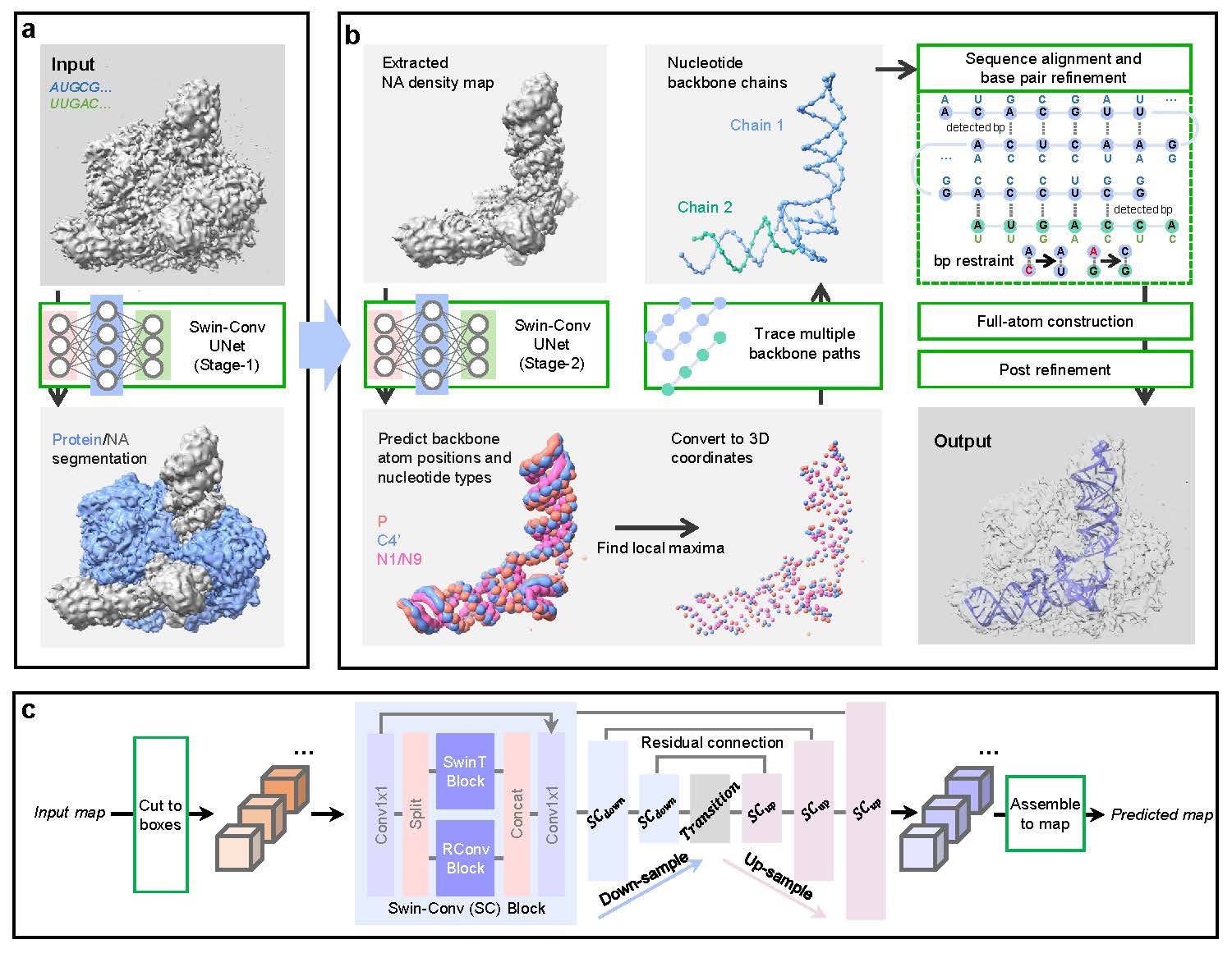
Figure 1: Workflow of EM2NA. a, EM2NA protein/NA segmentaion. b, EM2NA backbone atom detection and atomic modeling. c, Architecture of Swin-Conv-UNet (SCUNet).
1. Network architecture
A Swin-Conv-UNet (SCUNet) architecture is used to extract the DNA/RNA information from the map. The network consists of three encoder, one transition, and three decoder Swin-Conv (SC) blocks with skip connections between encoders and decoders. Here, the "Swin" represents the Shifted window transformer, which exhibits a good nonlocal modeling capability by computing self-attention of non-overlapping windows while also allowing for cross-window connection; The "Conv" stands for Convolutional network, which is known for it efficient local modeling capability. Therefore, compared with traditional networks like CNN, the Swin-Conv block has the advantage of both local and nonlocal learning capabilities. Each Swin-Conv block includes a Swin transformer (SwinT) block that is paralleled with a residual convolutional (RConv) block sandwiched by two 1x1 convolutions. The window size of the Swin transformer is set to 3. The 3D convolution layer with kernel size and stride of 2 is used as down-sampling, and the 3D transposed convolution layer with kernel size and stride of 2 is used as up-sampling. The inputs of our network are the density chunks of size 48x48x48 with a grid interval of 1.0 Angstrom. The outputs are of the same size. Our deep learning detection uses a two-stage pipeline to predict DNA/RNA backbone information. Both stage uses the same network but differs only in the final output channels.
2. EM2NA main pipeline
EM2NA first extract the NA-region from a raw input map by a first-stage SCUNet.
The NA-region is then input to the second-stage SCUNet to predict atom probability and nucleic acid identity for each grid.
With the predicted P, C4', and N1(N9) probability maps, the DNA/RNA backbone is constructed
by the following steps.
The predicted P and C4' atom probability maps are converted to the atom positions/points by detecting their local maximums using a mean-shift algorithm.
The combined P, C4' and N1/N9 positions are threaded to multiple chains by solving a Vehicle Routing Problem (VRP) algorithm.
The nucleotide types, i.e. A/G/C/U(T), can be assigned to the built DNA/RNA backbone chains/paths with or without providing DNA/RNA sequences.
If the target sequences are not provided, we will assign the nucleotide types solely with the deep learning-predicted ones.
Namely, the type (A/G/C/U(T)) for each nucleotide is assigned by voting the nearest voxels within 3 Angstrom of each C4' atom.
If the target sequence is provided, after the above assignment, a Smith-Waterman sequence alignment is used to inference the nucleotide types.
Based on the built DNA/RNA backbones and assigned nucleotide types, the full-atom structure is built by aligning template nucleotide conformations onto the backbone with the Kabsch algorithm.
If the target sequences are not provided, it is necessary to determine if the structure is an RNA(A-form) or DNA(B-form) according their helices.
Then, we can build the correct base conformation for the backbone.
To achieve this, we simply compare the detected pairings with the RNA/DNA helix templtes generated by 3DNA.
If more than 50% of the base pairs in two fragments are more similar to DNA(B-form), then the backbone fragments are built into the full-atom structure with DNA nucleotides, and vice versa.