Huang Lab
About EMBuild
EMBuild is a program for highly accurate model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembling

Copyright © 2021 Jiahua He, Peicong Lin, Ji Chen, Sheng-You Huang and Huazhong University of Science and Technology Released under GNU General Public License Version 3
EMBuild is freely available for academic or commercial users. If you have any questions regarding EMBuild, please don't hesitate to contact us at huangsy@hust.edu.cnDownload EMBuild
Click here to download EMBuild_v1.1
1. EMBuild_v1.1 is able to use the protein second structure annotations, which is predicted by a third program like EMInfo, through "-ssmap" option.
list of files EMBuild: the main program of EMBuild asgdom: program for labeling domains assigned by SWORD on PDB files rearrangepdb: program for rearrange the order of residues in PDB files EMBuild_single: single-thread version of EMBuild (Intel OpenMP is disabled) mcp/: UNet++ based main-chain probability prediction mcp/mcp-predict.py: the python script of main-chain probability map prediction mcp/frn.py: Pytorch implementation of Filter Response Normalization Layer used in main-chain probability prediction mcp/interp3d.f90: Fortran source code for interpolating EM grid mcp/model_state_dict: state dict of the trained main-chain prediction model mcp/model.py: Pytorch implementation of Nested U-net used in main-chain probability prediction mcp/utils.py: Python utilities used in main-chain probability prediction
Install the EMBuild
source /path/to/intel/oneapi/setvars.sh,
where /path/to/intel/oneapi/ stands for the local path of the installation directory of Intel® Fortran Compiler (as a part of Intel® oneAPI HPC Toolkit).
2. FFTW3 (http://fftw.org/)). Can be installed in Ubuntu via sudo apt-get install fftw3. Make sure libfftw3.so.3 is in LD_LIBRARY_PATH.
3. The maximum allowed stacksize of the system should be no less than 100MB. Use a large stacksize limit (in KB) by setting "ulimit -s". For example, set to 1GB (1048576 KB) by ulimit -s 1048576. If the stacksize limit is too small, EMBuild may encounter "Segmentation fault".
4. The SWORD program is used via a Perl script /path/to/SWORD/SWORD which combines multiple subprograms stored in /path/to/SWORD/bin/. However, the default path to /path/to/SWORD/bin/ in /path/to/SWORD/SWORD is just a relative path.Therefore, in order to use SWORD from other directories, users should change this path to the absolute path.
Specifically, there are six lines in the SWORD script /path/to/SWORD/SWORD that use the relative path: Line #7, Line #13, Line #16, Line #101, Line #384, and Line #475.
There is one line in /path/to/SWORD/bin/ParseMeasure.pmthat uses the relative path: Line #96.
There is one line in /path/to/SWORD/bin/ComputeJones/ComputeJones.pl that uses the relative path: Line #12.
How to Run EMBuild
Step 1: predicting main-chain probability map from EM density map
Instead of directly fitting protein chains to the original density map,
EMBuild fits the chains to the main-chain probability map predicted by deep learning.
Compared with the density map, the main-chain probability map includes more precise location information
of main-chain atoms, which can much help improve the accuracy of fitting.
In EMBuild, the main-chain probability map is predicted using
a nested U-net (UNet++) that was trained on a set of pairs of experimental density maps and main-chain probability
maps calculated from deposited PDB structures. The trained model can easily be applied to EM density maps through
mcp-predict.py.
In order to run python scripts properly, users should set the python path using one of the following ways:
1. Adding python path to the header of mcp-predict.py like "#!/path/to/your/python"
2. Running the scripts with the full python path like "/path/to/your/python mcp-predict.py -i IN_MAP -o OUT_MAP"
The interpolation program "interp3d.f90" should be built as a python package 'interp3d' through f2py. If the package cannot be imported properly on your system, please build it using f2py (the version of f2py should match the version of Python).
f2py -c interp3d.f90 -m interp3d
This command will generate an ELF file with name like "interp3d.cpython-*.so". Please keep "interp3d.cpython-*.so" with all python scripts "*.py" in the same directory.
Usage: mcp-predict.py -i in_map
-o out_map
-m model_state_dict_file
[Options]
Required arguments:
-i in_map: File name of input EM density map in MRC2014 format.
-o out_map: File name of the output main-chain probability map.
-m model_state_dict_file: State dictionary file for the parameters of the trained model (default is "./model_state_dict"). Users need to provide the path of "model_state_dict" if it is not in the working directory.
Options:
-g GPU_ID: ID(s) of GPU devices to use. e.g. "0" for GPU #0, and "2,3,6" for GPUs #2, #3, and #6. (default: 0)
-b BATCH_SIZE: Number of boxes input into the network in one batch. (default: 20)
-s STRIDE: The step of the sliding window for cutting the input map into overlapping boxes. Its value should be an integer within [10,40]. (default: 10)
--use_cpu: Whether to run on CPU instead of GPU.
Notes:
1. Users can specify a larger STRIDE of sliding window (default=10) to reduce the number of overlapping boxes to calculate. Since the size of the overlapping boxes is 40×40×40, the value of STRIDE should not exceed 40.
2. By default, main-chain probability prediction will run on GPU(s). Users can adjust the BATCH_SIZE according to the VRAM of their GPU. Empirically, an NVIDIA A100 with 40 GB VRAM can afford a BATCH_SIZE of 80. Users can run it on CPUs by setting --use_cpu. But this may take very long time for large density maps.
Example usage:
Predict main-chain probability map from the EM density map of EMD-0346.
The Python environment that satisfies all the package requirements is activated.
$ python3 mcp-predict.py -i ./emd_0346.map -o ./0346_MC.mrc -m ./model_state_dict
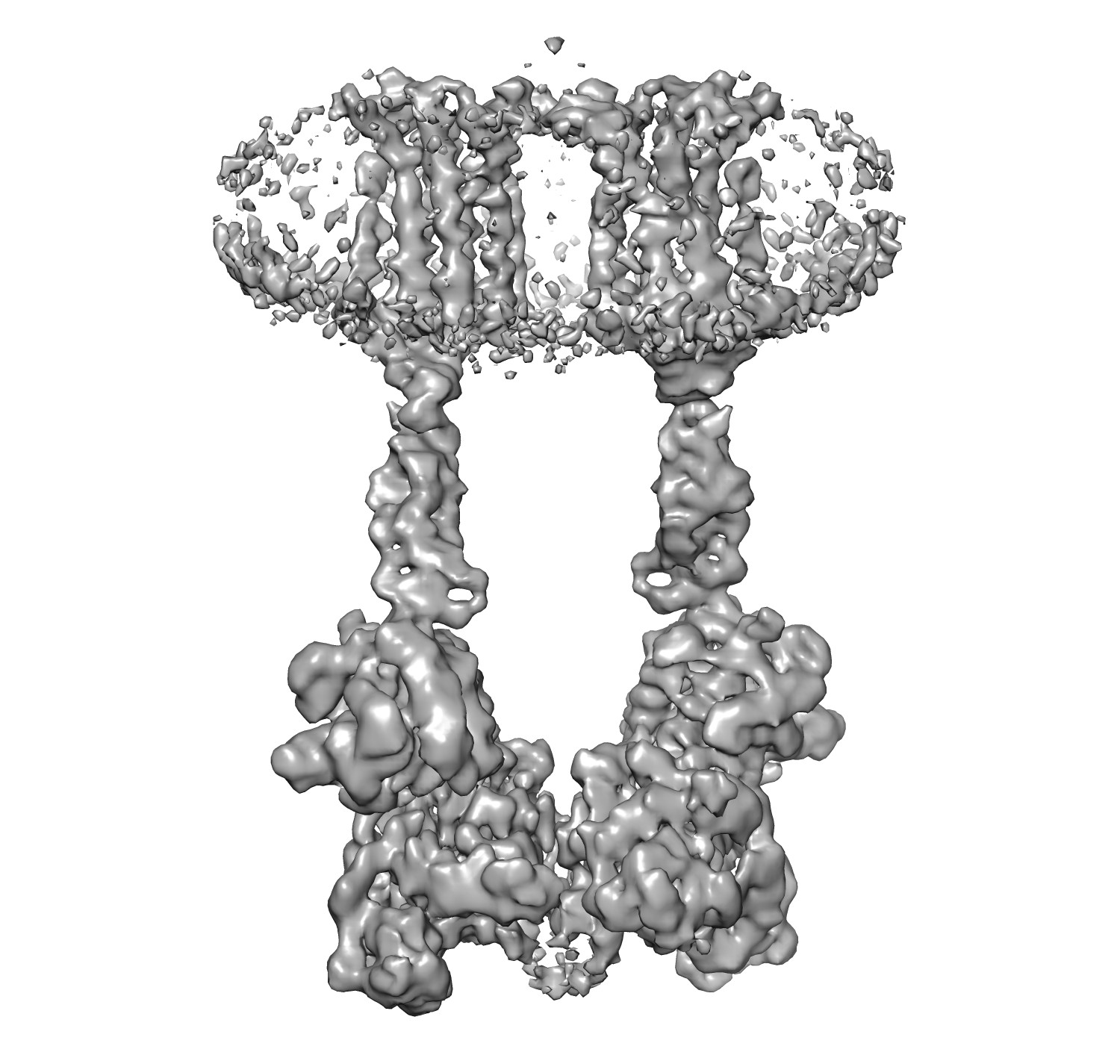
|

|
| "emd_0346.map" Input EM density map | "0346_MC.mrc" Output main-chain probability map |
Step 2: preparing the input chains for EMBuild
Given the input protein sequences of individual chains, their 3D structures are modeled by a protein structure prediction program.
In this work, AlphaFold2
is used to predict the protein structures from sequences, though other programs like
I-TASSER,
Rosetta,
and RoseTTafold could also be used.
For each predicted protein chain, we use
SWORD to assign its structural domains.
Note:
details about how to run
AlphaFold2 can be referred to
its original paper,
thus are not inculded in this tutorial.
During the evaluation of EMBuild, the native templates are excluded
by setting the "max_template_data" to the day before the
released date of the corresponding PDB structure of each test case,
in order to avoid bias in evaluation results.
However, in real applications,
users should skip this step to achieve the best modeling accuracy.
For each predicted chain, SWORD is used to assign its strutural domains, for example:
$ SWORD -i 6N52_A_AF2.pdb -m 15 -v > 6N52_A_AF2_SWORD.out
$ SWORD -i 6N52_B_AF2.pdb -m 15 -v > 6N52_B_AF2_SWORD.out
$ ./asgdom 6N52_A_AF2.pdb 6N52_A_AF2_SWORD.out 6N52_A_AF2_doms.pdb
$ ./asgdom 6N52_B_AF2.pdb 6N52_B_AF2_SWORD.out 6N52_B_AF2_doms.pdb
Note:
the SWORD assignment file should be generated from the input pdb.
The assigned domain IDs will be labeled in 71-74 columes of ATOM/HETATM records in the PDB file, as:
ATOM 2264 N LYS A 147 127.173 125.178 215.797 1.00 76.66 1 N
ATOM 2265 CA LYS A 147 127.292 124.277 214.649 1.00 76.66 1 C
ATOM 2266 C LYS A 147 127.521 125.098 213.370 1.00 76.66 1 C
ATOM 2267 CB LYS A 147 126.039 123.412 214.512 1.00 76.66 1 C
ATOM 2268 O LYS A 147 126.889 126.149 213.233 1.00 76.66 1 O
ATOM 2269 CG LYS A 147 125.867 122.403 215.655 1.00 76.66 1 C
ATOM 2270 CD LYS A 147 124.593 121.597 215.385 1.00 76.66 1 C
ATOM 2271 CE LYS A 147 124.343 120.522 216.441 1.00 76.66 1 C
ATOM 2272 NZ LYS A 147 123.097 119.800 216.093 1.00 76.66 1 N
ATOM 2273 N PRO A 148 128.034 126.129 212.743 1.00 84.85 2 N
ATOM 2274 CA PRO A 148 128.275 126.670 211.412 1.00 84.85 2 C
ATOM 2275 C PRO A 148 126.983 126.693 210.590 1.00 84.85 2 C
ATOM 2276 CB PRO A 148 129.328 125.754 210.779 1.00 84.85 2 C
ATOM 2277 O PRO A 148 126.108 125.832 210.749 1.00 84.85 2 O
ATOM 2278 CG PRO A 148 129.120 124.422 211.492 1.00 84.85 2 C
ATOM 2279 CD PRO A 148 128.719 124.853 212.901 1.00 84.85 2 C
REMARK LINK 147 148 1 2
REMARK LINK 200 201 2 5
REMARK LINK 339 340 5 3
REMARK LINK 470 469 5 4
REMARK LINK 517 518 5 6
REMARK LINK 397 398 3 4
REMARK LINK 574 575 6 7
REMARK LINK 644 645 7 8
REMARK LINK 781 782 8 9
TER
$ cat *AF2_doms.pdb > init_chains.pdb
Note: each chain should be ended with "TER" record.
Step 3: automatic modeling building with EMBuild
With the predicted main-chain probability map
and the predicted protein chains with domain assignments
generated respectively in Step 1 and
Step 2,
it is simple to apply EMBuild to automatically model the complex structure.
Usage: EMBuild
main-chain.map
input.pdb
resolution
output.pdb
[Options]
Required arguments:
main-chain.map: File name of the predicted main-chain probability map.
input.pdb: File name of the input PDB structure.
resolution: Resolution of the original EM density map in Angstroms.
output.pdb: File name of the output protein complex.
Options:
-nt N_THREADS: Number of threads/cores to run EMBuild. (default: 1)
--score:
Turn on scoring mode of EMBuild, the main-chain match score of input.pdb will be calculated in accordance to the provided main-chain probability map.
-stride STRIDE_PATH:
If scoring mode is turned on (--score), STRIDE_PATH should be provided,
in order to calculate the main-chain match scores of
continuous secondary structure fragments.
--help:
Print the detailed help message.
Notes:
1. Descriptions of some advanced parameters are not listed above, which can be shown using "--help" argument as $ ./EMBuild --help. It should virtually never be necessary to mess with the default parameters! Don't touch unless you know what you're doing!
2. There are two modes of the EMBuild program. One is the default mode that will model the complex structure from the input chains in input.pdb, for instance, modeling the structure of EMD-0346 at 4.0
Å resolution using 64 threads:
$ ./EMBuild 0346_MC.mrc init_chains.pdb 4.0 6N52_EMBuild.pdb -nt 64
$ ./rearrangepdb 6N52_EMBuild.pdb 6N52_EMBuild_rearranged.pdb
$ ./EMBuild 0346_MC.mrc 6N52_EMBuild_rearranged.pdb 4.0 6N52_EMBuild_scored.pdb \
--score -stride /path/to/stride
ulimit -s 1048576. If the stacksize limit is too small, EMBuild may encounter "Segmentation fault".
Example
$ cd ./6N52/
$ python ../mcp/mcp-predict.py -i emd_0346.map -o 0346_MC.mrc -m ../mcp/model_state_dict
$ SWORD -i 6N52_A_AF2.pdb -m 15 -v > 6N52_A_AF2_SWORD.out
$ SWORD -i 6N52_B_AF2.pdb -m 15 -v > 6N52_B_AF2_SWORD.out
$ ../asgdom 6N52_A_AF2.pdb 6N52_A_AF2_SWORD.out 6N52_A_AF2_doms.pdb
$ ../asgdom 6N52_B_AF2.pdb 6N52_B_AF2_SWORD.out 6N52_B_AF2_doms.pdb
$ cat *_doms.pdb > init_chains.pdb
$ ../EMBuild 0346_MC.mrc init_chains.pdb 4.0 6N52_EMBuild.pdb
$ ../rearrangepdb 6N52_EMBuild.pdb 6N52_EMBuild_rearranged.pdb
$ ../EMBuild 0346_MC.mrc 6N52_EMBuild_rearranged.pdb 4.0 6N52_EMBuild_scored.pdb \
--score -stride /path/to/stride
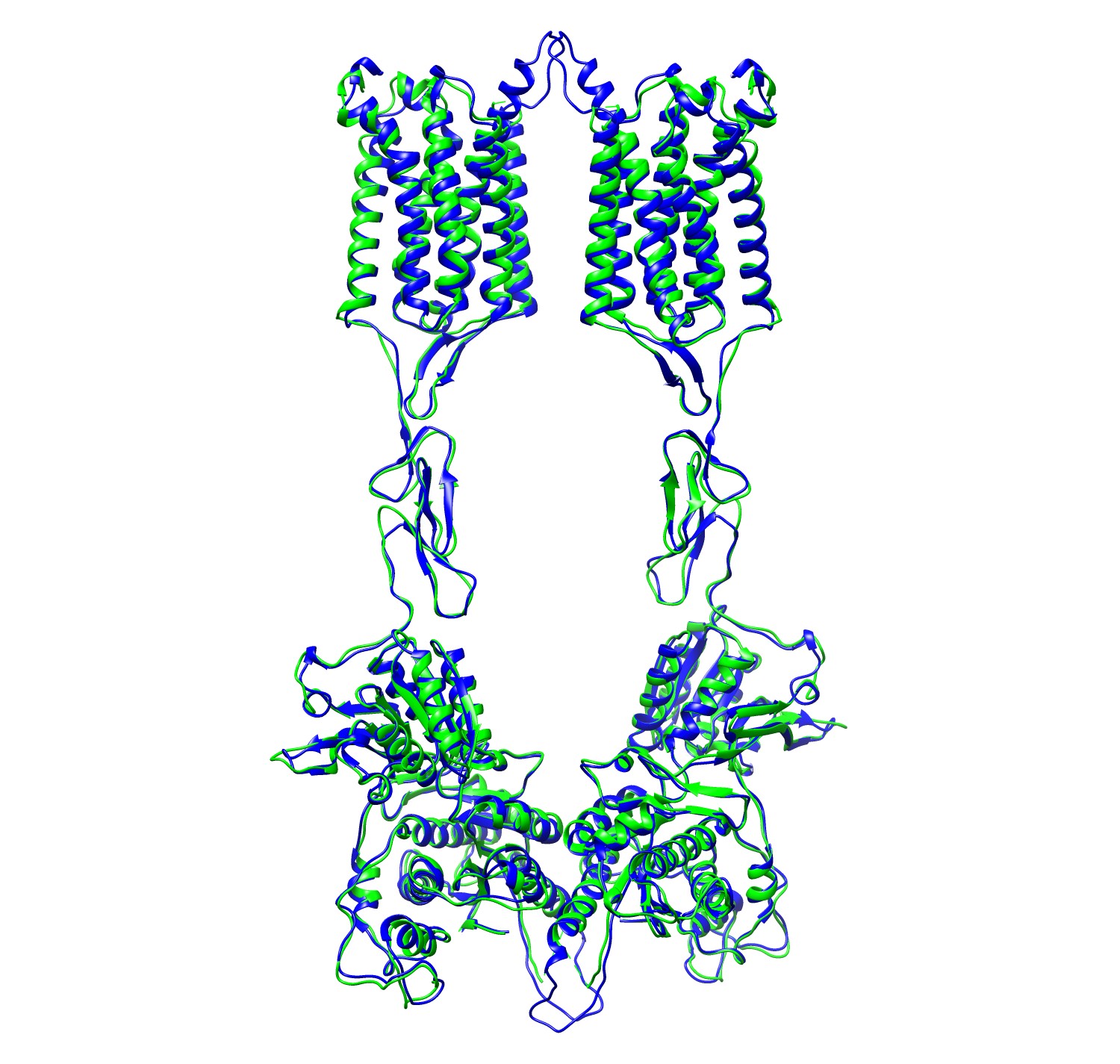 The PDB structure (6N52) is colored in green and
the EMBuild modeled structure is colored in blue
The PDB structure (6N52) is colored in green and
the EMBuild modeled structure is colored in blue
$ cd ./6PWP/
$ phenix.map_symmetry emd_20510.map symmetry=C7
$ python ../mcp/mcp-predict.py -i emd_20510.map -o 20510_MC.mrc -m ../mcp/model_state_dict
$ SWORD -i 6PWP_A_AF2.pdb -m 15 -v > 6PWP_A_AF2_SWORD.out
$ ../asgdom 6PWP_A_AF2.pdb 6PWP_A_AF2_SWORD.out 6PWP_A_AF2_doms.pdb
$ ../EMBuild 20510_MC.mrc 6PWP_A_AF2_doms.pdb 4.1 6PWP_EMBuild.pdb \
-fncs symmetry_from_map.ncs_spec
$ ../rearrangepdb 6PWP_EMBuild.pdb 6PWP_EMBuild_rearranged.pdb
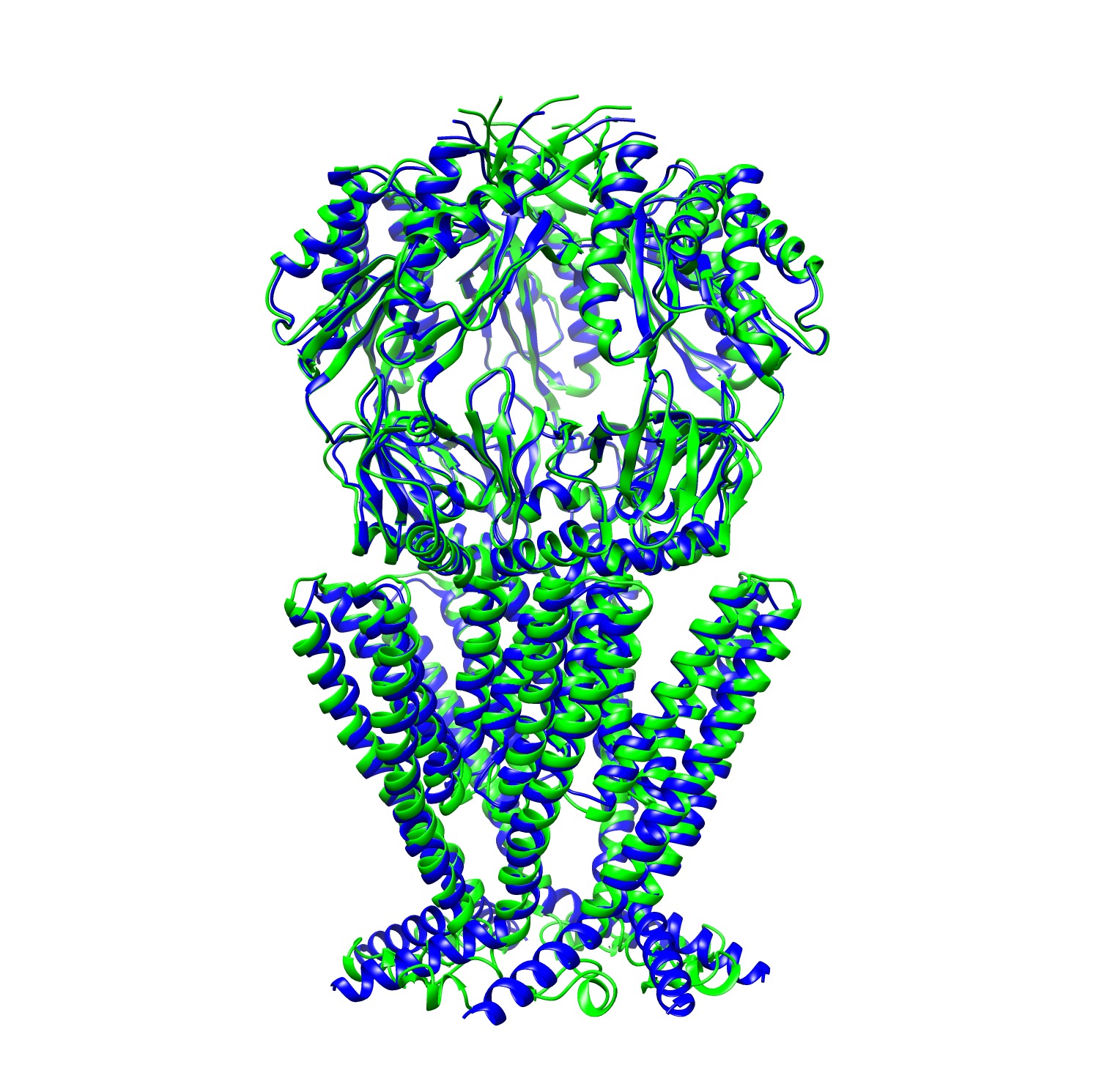 The PDB structure (6PWP) is colored in green and
the EMBuild modeled structure is colored in blue
The PDB structure (6PWP) is colored in green and
the EMBuild modeled structure is colored in blue